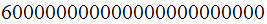 nucleotides.
nucleotides.
\begin{titlepage} \begin{center} %\vspace*{1.29375in} \vspace*{1.325in} {\Large Analysis on the accuracy of primary cancer detection\\using the Principal Component Analysis}\\ \vspace{.5in} Study using DNA Microarray Data for liver and bladder.\\ %\vspace{1.334375in} \vspace{1in} BY\\ \vspace{.1in} Nasser Abbasi\footnote{Student, Applied Mathematics Department, California State University, Fullerton.\newline A copy of this paper can be found at http://12000.org/index.htm}\\ \vspace{.2in} Final Project Report. Mathematics 501, Advanced Numerical Analysis\\ \vspace{.5in} Under supervision of Professor C.H. Lee\\ Department of Mathematics, California State University, Fullerton \end{center} \begin{abstract} Recent advances in microarray technology offer the ability to study the expression of thousands of genes simultaneously. The DNA data stored on these microarray chips can provide crucial information for early clinical cancer diagnosis. The Principal Component Analysis (PCA), also called the Principal Orthogonal Decomposition, has been widely used as an effective feature detection method. In this report, we implemented and applied PCA to study the accuracy of this method in detection of liver and bladder cancer data obtained from DNA microarray data. Our analysis discovered that the accuracy of PCA cancer detection can be improved by correlating the input sample whose cancer status is to be determined against a sample which represents the dominant gene expressions (hence forth called the eigensample) extracted by the use of PCA from the non-cancer samples as compared to the cancer samples. Additionaly, we analysed the effect on cancer detection accuracy of increasing the number of dominant eigensamples that we correlate the test sample against. We found that the first dominant eigensample is sufficient for this purpose. In addition, in the PCA training phase where feature extraction is performed, we analysed the affect of changing the number of the samples from which the eigensample is obtained (called the samples working set). We found that cancer detection accuracy remained relatively the same even when the samples working set was relatively small. \end{abstract} \end{titlepage}
Our approach is to apply a pattern recognition technique, called Principal Component Analysis (PCA) to detect the presence of primary cancer in human tissue by analyzing DNA microarray data that is published and available in public databases such as Stanford SMD and NCBI GEO.
PCA is used to extract the characteristics of a disease from an ensemble of samples known to carry the disease and to use the extracted feature for disease detection.
Such practice is quite common and was implemented using the Principal Component Analysis in [4, 5] in detecting cancers.
In this study, we analyzed the accuracy of PCA for cancer detection and found an approach which results in improved accuracy.
There are 2 main phases to the process of cancer detection using PCA. In the first phase, which is called the training phase, we use PCA to obtain the dominant eigensample which is then used in the second phase, which is called the detection phase by correlating the eigensample against the input sample whose cancer status is to be determined.
Finally, the accuracy of this result is determined by finding if the detection result was the correct prediction or not. This was possible to do in this study since the status of the input sample is known beforehand.
We applied the above approach in two different ways. First by obtaining the dominant eigensample from a random subset of cancer samples, and second by obtaining the dominant eigensample from a random subset of cancer free samples.
We then compared the accuracy of cancer detection in both case, and found that the accuracy is improved when the dominant eigensample is extracted from the cancer free set of samples.
The above analysis was performed on both liver and bladder cancer data. In both cases the accuracy result agreed. Using eigensamples from cancer free samples improves the accuracy of cancer detection.
Our findings indicate that this approach to the use of PCA for cancer detection provides a more accurate tool and that our approach can be a promising tool for clinical detecting of cancer as well as other diseases.
We start by a brief introduction to the biology involved, and then a description of the microarray data used. Then we present a mathematical introduction to PCA, followed by a description of the process used in this study in more details. Next we show the results found in tabular and graphical formats, and then give the study conclusions.
Biology is now starting to become an exciting field for applied mathematician and computer science since we can now apply many computational and mathematical analysis to its study thanks to the recent availability of DNA and genomic data.
There are more data currently being generated by biological sequencing (Both at the DNA and Protein levels) than there are computational tools and mathematical analysis available to analyze The large amount of data and complexity of biological interactions will require much more computational powers and smarter algorithms for its analysis than we can currently provide.
Mathematics can not only be applied to the analysis of DNA data, but also to the modeling of complex biological processes. For example, one very important areas in system biology is to better understand how gene expressions are regulated at the different molecular levels from DNA to mRNA to protein. Computational methods are developed to model this regulatory networks which involves complex mathematical models that range from discrete to continuous ordinary and partial differential equations as well as more advanced models involving stochastic processes.
I think that in this century, computation and mathematics will help greatly advance our understanding of biology as it similarly did to physics in the last century.
There is no better place to start this short review than with the central dogma of molecular biology.
The central dogma says that DNA goes to RNA which goes to protein. This is a one way street. Protein can never change to RNA, and RNA can never change to DNA. The process by which DNA becomes RNA is called transcription. The process by which RNA becomes protein is called translation.
RNA acts like the messenger for DNA, hence called mRNA. DNA tells mRNA to go make protein, and mRNA carries the information from DNA to make the specific protein. It is the type of protein made which determines the function and behavior of human cells.
Protein is the one which does all the work in the cell. One can think of DNA as the manager which makes orders and issues instructions to the cell, and mRNA is the messenger which builds a worker to do the actual work, and protein is the worker which mRNA made to do the work as instructed by DNA. Proteins themselves are also a long string of letters, but these letters are different from those than make DNA. Protein contain 20 different kinds letters, called amino acids.
DNA is the duplex helix strand that we are all familiar with, while RNA is a single strand DNA.
DNA is build of 4 basic chemical elements called nucleotides and go by the letters A,C,G, and T. Where A is adenosine, C is cytosine, G is guanine and T is thymine.
Since DNA is double stranded, then one strand will contain a string of these letters, and the second strand will contain a string of these letter as well, and these 2 strands are laid out on top of each others, where the letters from each strand approach the letters on the other strand and then they attach and lock themselves to each others, just like when the two parts of a zipper are closed and locked into each others.
The important thing to know is that the letter A will always attach itself to the letter T, while G attaches only to C. These pairs of bases are called complementary pairs.
DNA is the main central molecule from which all the cell functions originate.
Each human cell contain the same DNA letters. Each human cell contain about 3
billion base pairs. These are laid out in 46 different chromosomes. The
chromosomes are not all the same size, some are much longer than the others.
But the total base pairs in all of them is about 3 billion. Hence there are 6
billion nucleotides in each human cell. When a human cell divides and new cell
is created, a new 6 billion nucleotides are made in the process. The human
body contain large amount of cells, some estimate is at 100 trillion cells.
Hence the human body contain in it 100 trillion times 6 billion nucleotides or
nucleotides.
Back to DNA. We said that each cell contain about 3 billion base-pairs. However not all of this DNA is important. Only specific regions within the DNA are important. These regions are called genes. This is because this region of DNA is what gets transcribed to RNA and then translated to protein.
The human genome project and other studies estimate that there are about 30,000 genes in the human genome. This means out the 3 billion base-pairs in the cell DNA, there are only 30,000 locations where parts of the DNA is important. These are the genes. Each gene is of different length, where the length is measured in terms of base-pairs. Some genes can be thousands of bp (base pairs) long, while others can be only few hundreds long.
Since every human cell contain the same DNA data, why are cells different? The liver cell does things different from the bladder cell or the eye cell. The difference is that each of the above cells has different gene turned on. By having different genes turned on or off, the DNA will make different proteins. Proteins is what make cells different from each others.
How does cancer fit into this picture? Cancer occurs when human cell divides and duplicates without control. In a normal cell, protein controls the life cycle of a cell by controlling the production of new cells as old cells die. Since each specific protein is made by specific gene(s), knowing which genes are on or off in a cancerous cell, and how active that gene is gives us an indication of its role in the cancer which affected the cell.
Hence by obtaining a sample from a part of a body which is known to have cancer in it, and if by some process we are able to determine which genes are turned on , and how active these genes are in making protein and then compare this to a cancer-free sample of the same part of the body, we can then predict which genes contributed to the cancer and which did not.
For example, if we find that in all cancer sample, that gene XYZ was tuned on and is very busy (i.e. highly expressed) while in all cancer-free samples, from the same part of the body, that this same gene is either turned off, or if it is on, it is normally expressed (not as busy as in the cancerous samples). Then we can conclude that this gene has something to do with the cancer that affected this tissue. Further more, if we can find a group of genes which are always on together and are always highly expressed as a group in all cancerous samples, while the same group of genes are either not all on or not as highly expressed as a group, then we can conclude this group of genes work together in some fashion to cause the cancer.
How do we know which genes are more active than others? We do this by measuring the mRNA itself which is transcribed from that genes. In microarray technology, which we will describe shortly, it is able to measure the amount of specific gene mRNA. The more active a gene is, the more mRNA that the gene makes to produce more of the specific protein. The more mRNA is found in the sample, the more expressed a gene is, and this results on a stronger signal at that specific spot in the microarray as will be explained.
In a cancer experiment, detecting significant changes in a gene can indicate that this gene is a proto-oncogene which has turned into oncogene. Proto-oncogene are normal genes which are responsible for regulating cell growth. While an oncogene causes an increase in protein levels which are responsible for increased malignancy of tumor cell. Oncogene is a modified proto-oncogene resulting from mutation or other abnormalities which causes the proto-oncogene to turn to oncogene.
Some oncogene are involved in early stages of cancer, hence by having the ability to detect the presence of these types of genes can lead to early detection of cancer and hence can result in the ability to cure the cancer before it can spread and grow further.
Therefor, Knowing which genes are responsible for specific cancer can lead to a cure to that specific cancer by applying methods such as gene therapy or by using specific drugs which targets the proteins themselves.
For example, let us assume that we have 100 tissue samples which contain cancer and we see that in all of these sample that a some genes are turned on and are highly expressed, (meaning it is more active than other genes), and then compare this result to samples from the same tissue which are known to be cancer free. Then by doing this comparison, we are able to find which genes are more likely responsible for the cancer than others. This is where PCA comes into play. PCA allows us more easily to do this comparison by obtaining the signature signal of the cancer, and then by comparing against this signature of the cancer each new sample which makes cancer detection much simpler and more accurate.
Microarray technology is a recent invention to measure gene expressions. There are 2 types of microarray technology. spotted microarrays (or two-channel or two-colour microarrays), or oligonucleotide microarrays (or single-channel microarrays) (gene chips). The data used in this study used data from the spotted microarrays.
Microarrays measure mRNA in a sample. Let us concentrate on the microarray
used for this study. These microarrays contain
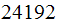 spots or positions on the microarray where each spot contain inside an
anchored probes which are oligonucleotides, a cDNA or small fragments of PCR
products corresponding to the specific mRNA to be detected. Only few base
pairs are needed to detect specific mRNA (12-14). The important thing for us
to know is that each spot will detect a specific mRNA, hence a specific gene
which transcribed this mRNA. How this happens in practice is that the mRNA
from the sample itself will attach itself to the correct spot by aligning its
bases to the bases of the probe in the microarray spot. Spots which gets
filled by mRNA will show up are more expressed than spots which are empty or
are not as expressed. By measuring the signal of the light from each spot, it
will give an indication of amount of mRNA that belong to that gene in the
sample.
spots or positions on the microarray where each spot contain inside an
anchored probes which are oligonucleotides, a cDNA or small fragments of PCR
products corresponding to the specific mRNA to be detected. Only few base
pairs are needed to detect specific mRNA (12-14). The important thing for us
to know is that each spot will detect a specific mRNA, hence a specific gene
which transcribed this mRNA. How this happens in practice is that the mRNA
from the sample itself will attach itself to the correct spot by aligning its
bases to the bases of the probe in the microarray spot. Spots which gets
filled by mRNA will show up are more expressed than spots which are empty or
are not as expressed. By measuring the signal of the light from each spot, it
will give an indication of amount of mRNA that belong to that gene in the
sample.
The important thing for the purpose of this study is to view each output from
a microarray experiment as sample which contains
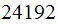 signals. Hence we have a vector of length
signals. Hence we have a vector of length
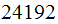 .
Each entry in this vector represent different gene.
.
Each entry in this vector represent different gene.
For the data used for the liver cancer analysis, the data contains a total of 207 samples. 76 of these are non-tumor, 105 are primary liver tumor, 74 are Metastatic tumor (This is tumor that primary in another part of the body but has spread to the liver), and 3 are Adenoma (benign lesions), 4 are benign tumor, and 10 are liver cell lines. Only sample that represent primary liver cancer and non-tumor samples are used. Hence a total of 105 tumor samples and 76 non-tumor samples are used for the liver cancer.